
-
News Message
基因编辑技术
- by wittx 2023-07-12
基因编辑技术是以特异性改变遗传物质靶向序列为目标的技术。近年来,锌指核酸酶(ZFN)、类转录激活因子效应核酸酶(TALEN)、规律成簇的间隔短回文重复(CRISPR)和单碱基编辑(BE)技术的相继出现,不仅为基因功能研究提供了有力的工具,还为生命医学提供了新的治疗方案。基因编辑技术已经大范围应用于动物细胞模型的构建、药物靶点的筛查和基因功能研究等,在基因治疗领域也展现出广阔的应用前景。本文就基因编辑技术的研究进展及其在基因治疗中的应用进行了概述,并对基因编辑技术的的原理、发展史、优缺点以及在基因治疗中的应用前景和机遇挑战进行了讨论,以期为基因编辑技术的临床转化提供参考。
关键词:基因编辑技术;CRISPR/Cas9;单碱基编辑;基因治疗

本文作者为:任云晓、肖茹丹、娄晓敏、方向东。发表于《遗传》杂志
基因编辑技术是以改变目的基因序列为目的,实现定点突变、插入或敲除的技术。从20世纪末人们就开始对基因编辑技术进行探索,但直到2013年CRISPR/Cas9技术成功用于哺乳动物细胞,才极大地推动了基因编辑技术的发展热潮。
真核生物的基因组包含数十亿个碱基,对其基因组的操作一直面临挑战。同源重组技术(homologous recombination, HR)是最早的基因编辑技术,也是真核生物基因编辑的一个重大突破。其原理是将外源性目的基因导入受体细胞,通过同源序列交换,使外源性DNA片段取代原位点上的基因,从而达到使特定基因失活或修复缺陷基因的目的。但是对高等真核生物来说,外源DNA与目的DNA自然重组率非常低,只有1/10E7~1/10E6;若要得到稳定遗传的纯合体基因敲除模型,至少需要两代遗传,因此HR的大规模应用受到了一定的限制。
为应对这一挑战,一系列基于核酸酶的基因编辑技术相继出现,实现了在真核生物尤其是哺乳动物中精准有效的基因编辑。与传统的基因编辑技术相比,基于核酸酶的基因编辑技术减少了外源基因随机插入,提高了对基因组特定片段进行精确修饰的几率。目前基因编辑技术主要包括以下几种:人工介导的锌指核酸酶技术(zinc finger nucleases, ZFNs)、类转录激活因子效应核酸酶技术(transcription activator- like effectors nucleases, TALENs)、规律成簇的间隔短回文重复相关蛋白技术(CRISPR/Cas9)和单碱基编辑(base editor, BE)技术等。
基因编辑技术掀起的研究热潮,一方面是因为基因编辑技术本身的发展,更为精准、高效、低成本的基因编辑技术不断地被开发出来;另一方面基因编辑技术作为一项重要的工具,在基因筛查、动物、细胞模型构建等基础研究中发挥着重要的作用,也为许多疾病的基因治疗提供了新的思路。因此,本文从基因编辑技术的发展历程及其在基因治疗中的探索和应用进行概述,并对基因编辑技术面临的挑战和机遇进行讨论。
1、基因编辑技术研究进展
基于DNA核酸酶的基因编辑技术发展迅速,从第一代DNA核酸酶编辑系统ZFNs、第二代TALENs到第三代CRISPR/Cas9系统,基因编辑效率不断提高、成本逐渐降低,应用范围不断扩大。ZFNs、TALENs和CRISPR/Cas9等3种基因编辑技术都是在基因组靶标位点引起DNA双链断裂(double-strand breaks, DSBs),进而激活细胞内部修复机制的基础上建立的。细胞内DNA双链断裂的修复机制包括易引起随机插入、缺失的异源末端连接(non-homologous end joining, NHEJ)和需要同源模板存在才可以激活的同源重组修复(homology directed repair, HDR)。2016年,BE技术的开发实现了在不引起DNA双链断裂和无需同源模板的情况下的单个碱基转换,有效地规避了基于双链DNA断裂后NHEJ和HDR修复的基因编辑技术的不足。
1.1 ZFNs
20世纪90年代,Fok I酶的发现促进了ZFNs的出现。1996年,美国约翰霍普金斯大学环境卫生科学系Chandrasegaran团队构建了基于Fok Ⅰ酶和锌指蛋白融合的ZFNs技术。ZFNs包含两个结构域:DNA结合的锌指蛋白区域和限制性核酸内切酶FokⅠ的核酸酶切活性区域(图1)。锌指蛋白区域决定了ZFNs的序列特异性。锌指基序一般由30个氨基酸组成,其结合锌离子的保守区域通常为4个半胱氨酸或2个半胱氨酸和2个组氨酸,其空间结构由1个α螺旋和2个反向的β平行结构组成。α螺旋的1、3、6位的氨基酸分别特异性地识别并结合DNA序列中的3个连续的碱基。由于不同的锌指基序中α螺旋的1、3、6位氨基酸不同,因此由3~6个不同锌指基序组成的锌指蛋白区域与FokⅠ核酸酶区域连接就构成了可以特异识别DNA序列并进行切割的人工核酸酶。FokⅠ必须二聚化才具有活性。由于FokⅠ自身二聚化也能对DNA进行切割,但是切割效率低且易产生非特异切割,所以在设计ZFNs时可以对FokⅠ进行突变,使之不能形成同源二聚体。当两个结合不同靶序列的突变的FokⅠ被5~7 bp的spacers隔开就可以形成具有核酸酶的活性的异源二聚体。这样设计的ZFNs可以增加其DNA序列识别的特异性。
基于Chandrasegaran的工作,美国犹他大学医学院生物化学系的Dana Carrol团队使用ZFNs注入果蝇胚胎,第一次实现了在动物中的基因编辑。随后,科学家用ZFNs技术在动物、植物和人类细胞中都实现了靶基因的编辑。尽管ZFNs技术在多个物种中成功进行了基因编辑,但是锌指蛋白的设计费时费力,成本较高,限制了该方法的大规模应用。
1.2 TALENs
2009年,美国爱荷华州立大学植物病理学与生物信息学系的Adam J. Bogdanove团队和德国马丁卢瑟大学生物研究所的Ulla Bonas团队分别发现了来自植物致病黄单胞菌属的转录激活效应蛋白(transcription-activator-like effector,TALE)和DNA的相互作用。将TALE蛋白与FokⅠ酶区域结合构建了新一代的核酸酶编辑技术——TALENs。TALENs的组成和ZFNs的相似之处是在其羧酸末端也含有FokⅠ核酸酶结构域,不同之处是TALENs的DNA结合域为TALE蛋白(图1)。TALE蛋白中每个识别模块由34个氨基酸组成,除了第12和13位氨基酸外其余氨基酸序列都是保守的,第12和13位氨基酸称为可变的双氨基酸残基(repeat variable di-residue, RVD)。RVD决定了TALE识别并结合的DNA碱基。4种不同的碱基都有与之对应的TALE识别模块。因此构建TALEN人工核酸酶时,只需要按照目标序列的顺序将不同的TALE识别模块的序列连接起来,再与FokⅠ的编码序列融合即可。相对于ZFNs来说,TALENs的设计变得容易,对任意的DNA序列理论上都可以设计和构建一个特异的TALEN核酸酶。但是目标序列的每个碱基都需要一个TALE识别模块,因此TALENs的构建过程工作量较大。此外,TALENs在人类细胞中的细胞毒性较低。2011年,Miller等第一次使用TALENs在人类细胞中对NTF3 和CCR5 基因进行编辑,证明了TALEN核酸酶对内源靶向基因的调节和修饰作用。
1.3 CRISPR/Cas9
2012年CRISPR/Cas9的体外重构和2013年在人类细胞中证明了其基因编辑功能,标志着新一代基因编辑时代的开始。CRISPR/Cas9系统来源于细菌和古细菌的天然获得性免疫系统,通过CRISPR RNA (crRNA)和trans-activating crRNA (tracrRNA)以及Cas9蛋白组成的复合体抵御外源性DNA的入侵。CRISPR/Cas9系统发挥作用的基本过程可以分为3个阶段:第一个阶段为间隔序列获得期,质粒或噬菌体携带的DNA片段被宿主的核酸酶切割成短的DNA片段,符合条件的DNA片段整合进宿主CRISPR位点成为crRNA重复序列间的间隔序列;第二个阶段为CRISPR/Cas9表达期,Cas9蛋白表达,CRISPR序列由pre-crRNA 加工为成熟的crRNA,成熟的crRNA包含间隔序列,靶向结合于外来入侵的DNA;第三阶段为DNA干扰期,Cas9蛋白在向导crRNA的引导下识别靶向位点,并调节基因组的剪切[22]。根据Cas蛋白的不同,CRISPR/Cas系统可以分为5类:类型Ⅰ、Ⅲ和Ⅳ的CRISPR位点包含crRNA与多个Cas蛋白形成的复合物;类型Ⅱ (Cas9)和类型V(Cpf1)只需要RNA介导的核酸酶。许多CRISPR系统都依赖于临近crRNA靶向位点的PAM (proto-spacer adjacent motif)序列,PAM序列的缺失将会导致Ⅰ型和Ⅱ型CRISPR系统的自我剪切。
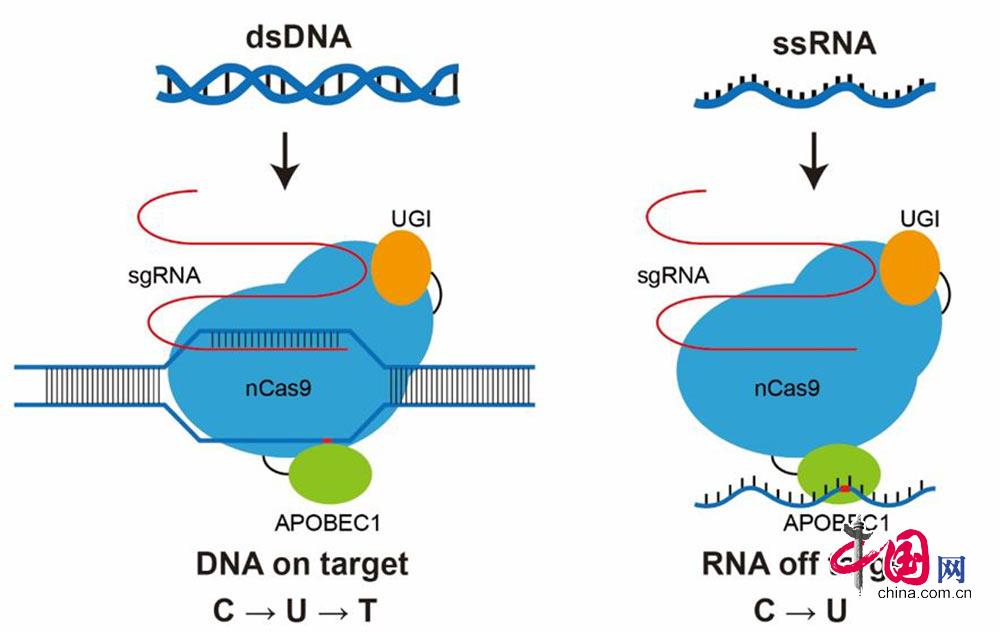
广泛用于基因编辑的CRISPR/Cas9是Ⅱ型CRISPR系统,由Cas9蛋白和sgRNA(single guide RNA)组成。sgRNA是根据crRNA和tracrRNA形成的高级结构设计的,与Cas9核酸酶蛋白结合,指导其识别并剪辑靶向序列,靶向序列附近必需存在含有NGG或者NAG的PAM基序(图1)。

图1
相比较ZFNs和TALENs,CRISPR/Cas9通过一段与目标DNA片段匹配的向导RNA引导核酸酶识别靶向位点,提高了Cas9核酸酶的特异性;同时,Cas9在sgRNA的引导下以单体蛋白的形式发挥功能,不像ZFNs和TALENs的FokⅠ酶只有二聚化才具有切割靶向DNA的活性,因此CRISPR/Cas9也避免了精细复杂的蛋白质设计或组装的需要。但是,由于CRISPR/Cas9来自于原核生物天然获得性免疫系统抵御外来遗传物质的防御系统,Cas9核酸酶可能继承了序列特异性低的特点,使其非特异性切割的几率增加,造成脱靶效应增多。不同的科研团队提出了不同的方法修饰或编辑Cas9和sgRNA以降低脱靶效应。如将Cas9蛋白与FokⅠ核酸酶、锌指蛋白或者TALE蛋白结合提高Cas9的特异性;用失活的Cas9蛋白和FokⅠ区域融合形成新的核酸酶,使其只有在核酸酶二聚化时才具有活性;Fatih等将锌指蛋白或者TALE蛋白与Cas9蛋白变异体融合增强核酸酶的特异性。另一种降低脱靶效应的方法是使用切口酶代替核酸酶,产生单链断裂而不是双链断裂,单链断裂不能诱导NHEJ的修复,仍然可以激活HR的精确修复。单链断裂可以使脱靶效应降低,但修复效率也降低,因此有人提出了使用双切口酶的方法,既提高了基因编辑的特异性也提高了编辑的效率。
1.4 BE
ZFNs、TALENs和CRISPR/Cas9技术都依赖于在靶位点诱导双链断裂进而激活DNA的NHEJ和HDR。NHEJ容易引起随机插入和缺失,造成移码突变,进而影响靶基因的功能;HDR尽管精确性高于NHEJ,但是其在细胞中的同源重组修复效率低,约为0.1%~5%。BE技术的出现有效地改善了以上问题。
2016年4月,美国哈佛大学David Liu实验室第一次发表了不需要DNA双链断裂也不需要同源模板即可进行单碱基转换的基因编辑技术——BE技术。该技术基于无核酸酶活性的dCas9 (Inactive, or dead Cas9)或有单链DNA切口酶活性的Cas9n (Cas9 nickase)、胞嘧啶脱氨酶、尿嘧啶糖基化酶抑制子(uracil DNA glycosylase inhibitor, UGI)以及sgRNA形成的复合体,在不引起双链DNA断裂的情况下,直接使靶向位点的胞嘧啶(Cytosine, C)脱氨基变成尿嘧啶(Uracil, U);由于尿嘧啶糖基化酶抑制子的存在,抑制了U的切除;随着DNA复制,U被胸腺嘧啶(Thymine, T)取代;同时,互补链上原来与 C 互补的鸟嘌呤(Guanine, G)将会替换为腺嘌呤(Adenine, A),最终实现了在一定的活性窗口内C 到 T 和G 到A 的单碱基精准编辑。BE 技术的出现促进了点突变基因编辑的有效性和使用范围。
David Liu 团队对4 种胞嘧啶脱氨酶——hAID(human activation induced deaminase)、hAPOBEC3G(human apoliprotein B mRNA-editing enzyme-catalytic polypeptide-like-3G)、rAPOBEC1 (rat apolipoprotein B mRNA-editing enzyme 1)和七鳃鳗(Lethenteron camtschaticum)来源的AID 类似物PmCDA1 进行评估, 发现rAPOBEC1 具有最高的脱氨酶活性。他们通过将rAPOBEC1 与dCas9 的N 末端以及16 个残基的XTEN 连接体融合,组成了第一代碱基编辑器BE1 (rAPOBEC1-XTEN- dCas9),具有5 nt 的活性窗口。第二代碱基编辑器BE2 (APOBEC-XTENdCas9-UGI)融入了UGI 抑制了U 糖基化引起碱基切除修复,编辑效率在人类细胞比BE1 高3 倍。第三代碱基编辑器BE3 (APOBEC-XTEN-dCas9(A840H)-UGI)恢复了Cas9 HNH 区域840 位置组氨酸的催化作用,可以剪切非编辑链上与编辑链上U 互补的G碱基,对非编辑链的剪切使BE3 的编辑效率比BE2高2~6 倍。
随着单碱基编辑技术的出现,日本科学家基于 PmCDA1 和dCas9 或Cas9n 以及鸟嘌呤糖苷化酶抑 制子UGI 的融合,也开发了PMCDATA-dCas9/Cas9n-UGI 的单碱基编辑器。中国科学院上海营养与健康研究院常兴研究组开发了基于hAID 的dCas9-AIDx 单碱基编辑系统,用于耐药突变的筛选。
2017 年,David Liu 实验室对BE 技术做了多方面改进。首先针对常用的化脓性链球菌(Streptococcus pyogenes)的Cas9(SpCas9)蛋白靶向范围较窄,只识别含有NGG或NGA的PAM序列的靶位点的限制,他们使用金黄色葡萄球菌(Staphylococcus Aureus)的Cas9 (SaCas9)、SaCas9 突变体(Sacas9-KKH)、SpCas9突变体(SpCas9-VQR、SpCas9-EQR、SpCas9-VRER)替代SpCas9,可以识别含有NGG、NGA、NGAN、NGAG、NGGG、NNGRRT 和NNNRRT 的PAM 序列,从而显著提高了单碱基基因编辑的靶向范围。其次,他们通过突变胞嘧啶脱氨酶,降低酶的活性、改变底物的结合和构象,或者直接降低底物进入胞嘧啶脱氨酶活性区域的能力,缩小了单碱基编辑系统的活性窗口,从5 个核苷酸窗口缩小至1~2 个核苷酸活性窗口。再次,David Liu 实验室通过对BE3引入突变来减少脱靶效应,产生了高保真的碱基编辑器(high-fidelity base editor, HF-BE3)。随后,他们又报道了基于E. coli TadA (ecTadA)的新型单碱基编辑器——腺嘌呤碱基编辑器(adenine base editors, ABEs),实现了A⋅T 碱基对向G⋅C 碱基对的转换。经过不断的改进,第7 代的腺嘌呤碱基编辑器将靶向的A.T碱基转化为G⋅C 碱基对可以达到约50%的效率(人类细胞),并且引入插入或缺失的频率低于0.1%。

2、基因编辑技术在基因治疗中的应用
从 1996 年对ZFNs 第一次进行体外验证到2012年CRISPR/Cas9 技术的出现和之后的蓬勃发展(图2),基因编辑技术发展迅速,编辑效率和精确性不断提高,应用领域也不断拓宽。不仅可用于表达调控和基因功能的研究、细胞动物模型的构建、癌基因和药物靶点的筛选,在基因治疗中更是具有巨大的发展前景,为单基因遗传病、癌症等疾病提供了新的治疗方法。

图2
基因治疗通过导入正常基因或者编辑修复缺陷基因,实现治疗疾病的目的。目前,利用基因编辑技术在多种疾病,如单基因遗传病、眼科疾病、艾滋病及肿瘤等的基因治疗中得到了应用。
镰状细胞病(sickle cell disease, SCD)是由于β-珠蛋白基因的第7 个密码子的单基因点突变造成的。Hoban 等利用ZFNs 特异性靶向于β-珠蛋白基因,并诱导CD34+造血干细胞和祖细胞中的DNA 被切割。当ZFNs 与整合酶缺陷型慢病毒载体或寡核苷酸供体一起传送进细胞时,有效地实现了β-珠蛋白基因座的基因矫正。Hoban 的研究为镰状型细胞性贫血的基因治疗提供了重要的方法路径。
基因编辑技术不仅应用于遗传性疾病的治疗,在非遗传性疾病中也有重大的突破。与年龄相关的黄斑衰退(age-related macular degeneration, AMD)是导致成人失明的主要原因,脉络膜新血管形成(choroidal neovascularization, CNV)是其主要病理学特征,血管生成因子如VEGFA (vascular endothelial growth factor A, VEGFA)基因的高表达是造成病变的主要原因。Kim 等[47]将预先设计好的Cas9 核糖核蛋白 (ribonucleoprotein, RNP)导入成年小鼠眼中,使视网膜色素上皮中VEGFA 基因失活;并且在AMD 的小鼠模型中发现cas9 RNPs 有效地减少了脉络膜新血管生成的面积。该研究表明CRISPR/Cas9 技术有可能用于非遗传性退行性眼部疾病的局部治疗。
基因编辑技术在艾滋病、肿瘤治疗等领域也取得了初步进展。第一次在人类中应用靶向核酸酶是利用ZFNs 技术编辑CCR5 基因抵抗HIV。研究者从HIV 病人中提取T 细胞,用ZFNs 技术干扰T 细胞中CCR5 基因,由于CCR5 基因是大部分HIV 菌株尤其是早期感染菌株的辅助受体,干扰CCR5 基因的表达可以抗HIV 感染,表明基因编辑技术可能成为艾滋病治疗的新方向。
基因编辑应用于肿瘤治疗,主要是与免疫治疗相结合,尤其是与CAR (chimeric antigen receptor) T细胞,该方法在白血病、淋巴瘤和部分实体瘤中有巨大的发展前景。CARs 包括肿瘤细胞特异抗原的胞外单链可变片段和细胞内嵌合信号域,可以激活T 细胞和杀伤肿瘤细胞。Ren 等[54]使用CRISPR/Cas9 系统同时破坏多个基因位点,产生的TCR (T cell receptor)和HLA-I (HLA class I)缺陷的CAR-T细胞可作为通用的CAR-T 细胞,用于免疫治疗;除了产生通用的CAR-T 细胞,基因编辑技术也可通过敲除编码T 细胞抑制受体或信号分子的基因如PD1(programmed cell death protein 1)和CTLA4 (cytotoxic T lymphocyte-associated protein 4),用于产生增强型CAR-T 细胞。
2016 年,四川大学华西医院卢铀团队开展了CRISPR 基因编辑技术的临床实验,从转移性非小细胞肺癌患者中分离出T 细胞,并使用CRISPR/Cas9 技术敲除细胞中的PD-1 基因,在体外扩增到一定量后再重新输回患者体内,达到杀死肿瘤细胞的目的但是简单的敲除T细胞的抑制因子是一把双刃剑,真正投入临床使用还需要进一步研究敲除这些抑制因子是否会引起细胞的不可控制的增殖或者产生严重的自身免疫。
由于BE技术是在不造成双链DNA断裂的情况下进行的精确碱基转换,这对于基因治疗而言无疑是一个非常有效的工具。β-地中海贫血是由于珠蛋白基因(hemoglobin-beta, HBB)突变所致,中国和东南亚地区的发病原因主要是HBB基因A到G的突变。2017年,中山大学黄军就团队利用单碱基编辑技术在不能发育成熟的人类三元核胚胎中战对HBB的点突变进行编辑,即将碱基G改为A从而修正错误。该研究是第一个利用BE技术对遗传疾病突变位点进行精准修复的研究,为治疗新生儿β-型地中海贫血症,甚至为其他遗传性疾病的治疗打开了新窗口。Chadwick团队也通过BE技术实现PCSK9基因的敲除,从而降低血浆胆固醇的水平。
3、基因编辑技术面临的挑战
尽管基因编辑技术在基因治疗领域展现了广阔的应用前景,但是目前仍然面临着诸多挑战,如脱靶效应、传递系统的有效性和安全性、免疫排斥反应、伦理争论等。
3.1 脱靶效应
脱靶效应会导致假表型,造成错误的理解和解读,是限制基因编辑技术应用的重要原因。脱靶效应面临两个问题:一是如何从技术本身上降低脱靶效应;二是如何提高检测方法的灵敏性。降低脱靶效应的可行性策略有减少NHEJ修复引入的插入和缺失,如使用单切口酶活性突变体和不需要双链断裂激活修复的BE技术等;改进sgRNA的设计,如在保证一定的sgRNA结合靶点效率的基础上,截短sgRNA的长度;提高核酸酶蛋白的特异性,如David Liu研究组通过在Cas9蛋白特定位点插入羟基他莫昔芬应激性内含肽(hydroxytamoxifen (4-HT)- responsive intein)产生小分子激活Cas9核酸酶,使Cas9靶向编辑的特异性提高了25倍。
目前脱靶效应的检测方法有基于全基因组范围内检测DSBs的方法,该方法可以无偏向性地评估Cas9剪切的特异性,如通过NHEJ将双链寡聚脱氧核苷酸整入DSBs,进而扩增和测序的GUIDE-Seq (genome-wide unbiased identification of DSBs enabled by sequencing),以及用Cas9体外消化分离基因组DNA,然后进行全基因组测序的Digenome-Seq (digested genome sequencing)。虽然两种方法都无偏向性,且敏感性较好,但是两者都需要参考基因组,GUIDE-Seq整合双链寡聚脱氧核苷酸进入DSBs的效率不高,Digenome-Seq检验gRNA费用昂贵,且检测多个gRNA时测序深度也受限制。因此,脱靶效应仍然是基因编辑技术未来发展必需解决的问题之一。
3.2 核酸酶的传递效率和安全性
核酸酶的传送系统包括病毒、质粒、RNA和蛋白质的系统,其中病毒载体系统具有较高的编辑效率和插入突变,较低的脱靶效应和较高的免疫反应,如AAV (adeno-associated virus)载体和慢病毒载体广泛用于靶向基因向细胞的传送,但是其对传输物质大小有限制;质粒型载体在编辑效率、插入突变、脱靶效应和免疫反应均具有中等表现;RNA或蛋白质核酸复合物均不具有插入突变的功能,但普遍认为以蛋白核酸复合物进行传输具有起效快且无长期表达的特点,因而具有较低的脱靶效应。就BE系统来说,需要Cas9与胞嘧啶脱氨酶、UGI、sgRNA的融合,序列组成较长。如果使用AAV载体,其对包装物质4.7 kb长度的限制,使BE系统无法被包装进去。可以考虑将BE系统拆分包装成两个病毒,但是系统拆分后的编辑效率是否会受到影响尚不得知。因此,提高核酸酶传送系统的有效性和安全性将会进一步促进基因编辑的应用,有研究表明脂质体可能在基因编辑传送系统中展现巨大的发展潜力。
3.3 基因编辑引起的副作用
美国斯坦福大学儿科血液学家Matthew Porteus和Kenneth Weinberg的研究小组发现人体中普遍存在Cas9抗体,如果使用CIRSPR/Cas9进行治疗,可能引发人体剧烈的免疫反应,最终导致治疗失败,但是目前有关基因编辑可能引起免疫反应的研究还相对较少。
近期,Nature Medicine发表了分别来自瑞典卡洛林斯卡研究所和美国剑桥诺华生物医学研究院的研究。他们分别独立发现CRISPR/Cas9基因编辑过程造成的DNA双链断裂可以激活p53通路。这意味着基因编辑成功的细胞很可能成为潜在的癌细胞,利用CRISPR/Cas9进行临床治疗可能无意中增加患癌症的风险。
CRISPR/Cas9技术除了面临脱靶问题,可能还面临染色体结构异常的问题。Kosicki等在Nature Biotechnology上报道了Cas9还可能在作用靶点附近导致大规模的DNA删除,在部分情况下,甚至引起复杂的DNA重排。研究人员通过长读长测序和大范围的PCR基因型鉴定发现在小鼠胚胎干细胞和人类造血祖系细胞内,可能出现数千DNA碱基的删除,导致临近基因或调控序列受到影响并改变细胞功能。因此引起的DNA重排问题也成为了CRISPR/Cas9技术面临的另一个安全隐患。
除了上述挑战之外,基因编辑技术在基因治疗中的应用仍可能有很多意想不到的问题出现。但无论如何,技术的发展总会遇到瓶颈,瓶颈的攻克也将必然给技术的应用带来更广阔的空间。
3.4 基因编辑中的伦理问题
基因编辑技术为遗传性疾病、肿瘤等多种疾病带来了治疗的希望,但是人类胚胎细胞的基因编辑引起了社会上广泛的讨论和激烈的争议。一方面,对胚胎细胞的基因编辑有助于基础科学研究,促进我们了解人类胚胎发育的分子机制,有利于实现在早期胚胎发育时期治愈疾病的目标;另一方面基因编辑技术可能产生的脱靶或者其他基因编辑失误,从而导致慢性疾病或者严重的致残个体,这些对人类的繁衍和生活方式等都将会产生非常大的危害。然而,目前很多国家的法律对基因编辑技术应用于基因治疗的监管措施依然存在大量空白。
4、结语与展望
基因编辑技术的快速发展极大地提高了我们对真核细胞基因组进行精准改变的能力。可编辑的核酸酶,尤其是编辑效率更高、操作简单、成本低的CRISPR/Cas系统,彻底改变了我们对基因组功能的研究;单碱基编辑技术的出现和不断改进,实现了单个碱基的精准转换,降低了脱靶效率。虽然目前基因编辑技术仍然面临着脱靶效率和潜在的免疫反应等副作用的问题,相信在未来多学科的交叉融合和科学家的共同合作下,新一代基因编辑技术将更加简便、高效、精确,目前存在的问题也会逐步得到解决。随着人们对疾病的新的有效靶点的不断认识和新的基因编辑技术的不断发展,基因编辑治疗方案的大规模临床应用将成为可能,特别是对那些传统疗法难以治愈的疾病,可以通过纠正致病基因或引入有益突变来达到治愈疾病的目的。基因编辑技术的临床转化和应用研究值得拭目以待,这将成为下一代转化疗法和治疗范例的关键,也将促进个体化医疗的快速发展。
Share Http URL: http://www.wittx.cn/get_news_message.do?new_id=1199

Best Last Month

Information industry by wittx
Information industry by wittx
Information industry by wittxVision Transformer Advanced by Exploring Inductive Bias for Image Recognition and Beyond

Information industry by wittx
Information industry by wittx
Information industry by wittxEmpowering Transformers for Times Series Forecasting with Exogenous Variables
.jpg)
Information industry by wittx
Information industry by wittx
Information industry by wittx

Information industry by wittx
